Usage
Usage modes
MS²PIP has various usage modes that each can be accessed through the command-line interface, or through the Python API.
predict-single
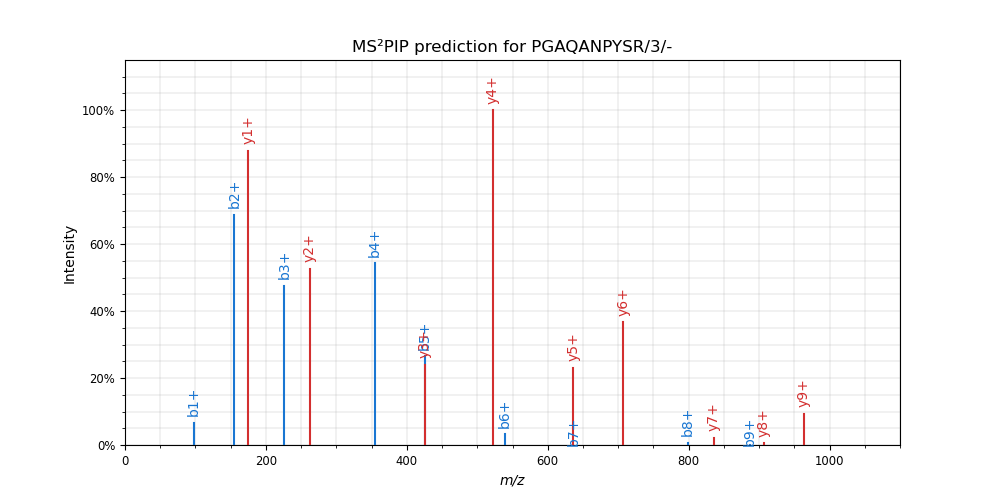
In this mode, a single peptide spectrum is predicted with MS²PIP and optionally plotted with spectrum_utils. For instance,
ms2pip predict-single "PGAQANPYSR/3" --model TMT --plot
results in:
predict-batch
Provide a list of peptidoforms (see Peptides / PSMs) to predict multiple spectra at once. For instance,
ms2pip predict-batch peptides.tsv --model TMT
results in a file peptides_predictions.csv with the predicted spectra.
predict-library
Predict spectra for a full peptide search space generated from a protein FASTA file. Various
peptide search space parameters can be configured to control the peptidoforms that are generated.
See ms2pip.search_space for more information.
Minimal example:
ms2pip predict-library proteins.fasta
This mode was first developed in collaboration with the ProGenTomics group for the MS²PIP for DIA project.
correlate
Predict spectrum intensities for a list of peptides and correlate them with observed intensities from a spectrum file. This mode is useful for evaluating MS²PIP models or for (re)scoring peptide-spectrum matches.
For instance:
ms2pip correlate --psm-filetype sage results.sage.tsv spectra.mgf
correlate-preloaded
Compare predicted and observed intensities for PSMs that already have
ms2rescore_rs.MS2Spectrum objects attached to their spectrum attribute. This is
useful when spectra are already loaded in memory, e.g., when using MS²PIP within
ms2rescore. This mode is only available through the
Python API.
import ms2pip
results = ms2pip.correlate_preloaded(psm_list, compute_correlations=True)
correlate-single
Predict spectrum intensities for a single peptide and correlate them with observed intensities from
an ObservedSpectrum object. This mode is only available through the Python API, not
through the command-line interface.
get-training-data
Given a list of peptides and corresponding spectra, generate training data for MS²PIP. This includes observed intensities for the supported ion types and the feature vectors for each ion. For more info, see Training new MS²PIP models.
annotate-spectra
Given a list of peptides annotate the peaks in the corresponding spectra.
Input
Peptides / PSMs
PSM file types
For peptide information input, MS²PIP accepts any file format that is supported by
psm_utils. See
Supported file formats for
the full list. The simplest format is a tab-separated file with at least the columns
peptidoform and spectrum_id present.
peptidoformis the full ProForma 2.0 notation including amino acid modifications and precursor charge state.spectrum_idshould match theTITLEornativeIDfield of the related spectrum in the optional MGF or mzML file, if provided. Otherwise, any value is accepted.
For example:
peptidoform spectrum_id
RNVIM[Oxidation]DKVAK/2 1
KHLEQHPK/2 2
...
See psm_utils.io.tsv for the full specification.
Peptide sequence properties
Peptides must be strictly longer than 2 and shorter than 100 amino acids and cannot contain the following amino acid one-letter codes: B, J, O, U, X or Z. Peptides not fulfilling these requirements will be filtered out and will not be reported in the output.
Amino acid modifications
Amino acid modification labels must be resolvable to a known mass shift. This means that accepted labels are:
A name or accession from an controlled vocabulary, such as Unimod or PSI-MOD. (e.g.,
Oxidation,U:Oxidation,U:35,MOD:00046…)An elemental formula (e.g,
Formula:C12H20O2)A mass shift in Da (e.g.,
+15.9949)
Any unresolvable modification will result in an error. If needed, PSM files can be converted with
psm_utils.io and modifications can be renamed with the
rename_modifications() method.
Spectrum file
In the correlate and get-training-data usage modes, a spectrum file with observed spectra must be provided to MS²PIP. Spectrum files in mzML, MGF, Thermo raw, and Bruker raw formats are supported.
Note
To read Thermo raw files, the .NET runtime must be installed.
Make sure that the PSM file spectrum_id matches the MGF TITLE field or mzML nativeID
fields. If the values of these fields are different, but the PSM file spectrum_id is embedded
in them, the spectrum_id_pattern argument can be used to extract the spectrum_id from
the TITLE or nativeID fields with a regular expression pattern. For example, if an MGF
entry has TITLE=scan=1, but the PSM file has spectrum_id=1, spectrum_id_pattern can be
set to scan=(\d+). Note that the pattern must contain a single matching group that captures the
spectrum_id.
Note
Find out more about regular expression patterns and try them on regex101.com. You can try out the above examples at https://regex101.com/r/TynuIe/1.
Spectra present in the spectrum file, but missing in the PSM file (and vice versa) will be skipped.
Output
MS²PIP supports various spectral library output formats, including TSV, MGF, MSP, Spectronaut CSV, BiblioSpec/Skyline SSL and MS2, and Encycopedia DLIB.
Note that the normalization of intensities depends on the output file format. In the TSV file output, intensities are log2-transformed. To “unlog” the intensities, use the following formula:
intensity = (2 ** log2_intensity) - 0.001